AAV在腦組織中的靶向策略(應(yīng)用篇)
· 案例一 ·
“TNF-α-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG”
阿爾茨海默病(AD)是一種常見的神經(jīng)退行性疾病,臨床表現(xiàn)為認(rèn)知功能障礙和記憶力減退等����,其病理特征包括Aβ蛋白沉積�、Tau蛋白過度磷酸化、神經(jīng)炎癥和神經(jīng)元壞死等�。研究表明,AD發(fā)病的病理過程可能與神經(jīng)元壞死和自噬有關(guān)���。神經(jīng)元壞死是AD的主要特征�����,程序性細(xì)胞壞死與AD的發(fā)病機(jī)制相關(guān)��,然而AD中誘導(dǎo)神經(jīng)元壞死的機(jī)制尚不清楚。自噬受損導(dǎo)致Tau蛋白的積累����,這是包括AD在內(nèi)的大多數(shù)神經(jīng)退行性疾?���。∟Ds)的特征��。然而���,目前對(duì)AD發(fā)病機(jī)制中自噬途徑和神經(jīng)元壞死之間可能的相互作用知之甚少�。TNF-α作為關(guān)鍵促炎因子,其誘導(dǎo)的炎癥可能對(duì)神經(jīng)元死亡產(chǎn)生不利影響���,但在AD發(fā)病過程中TNF-α的升高是否能引起神經(jīng)元壞死還尚未可知。
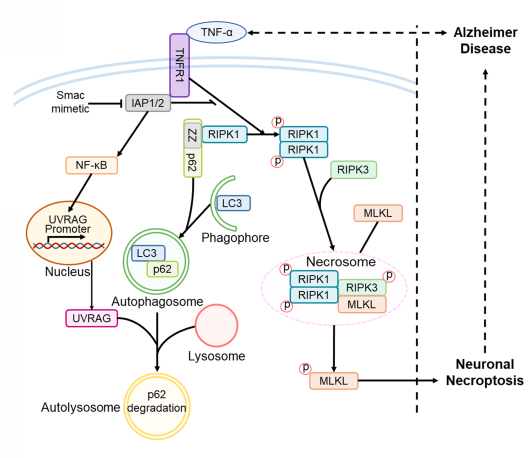
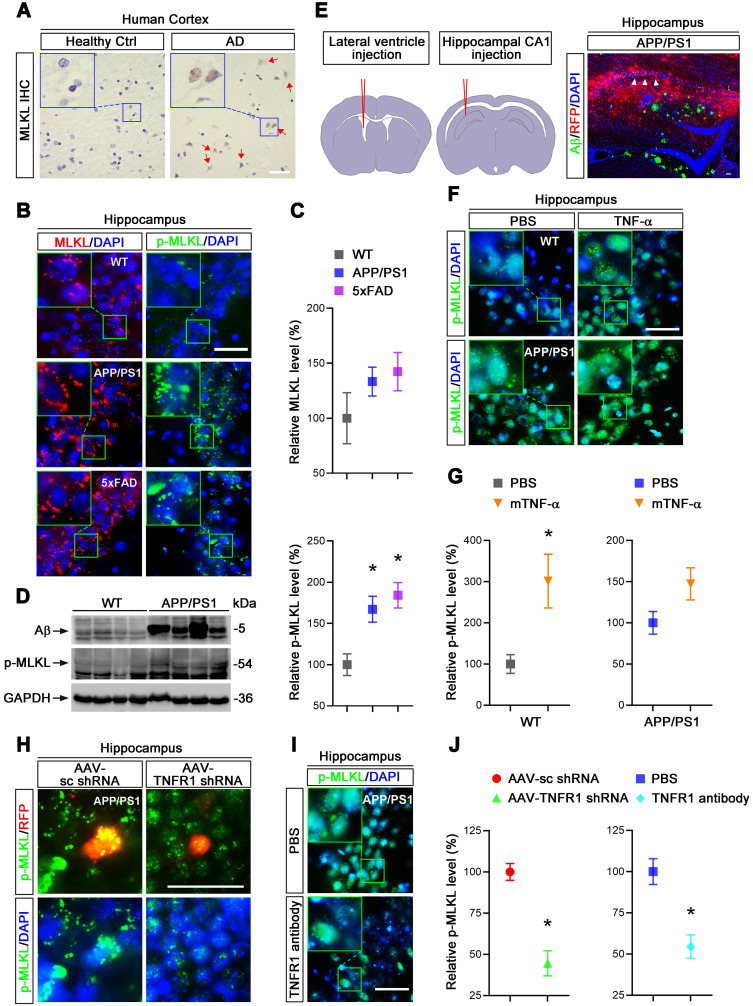
圖1. 阿爾茨海默病中神經(jīng)元壞死的調(diào)控機(jī)制
本研究利用來自AD患者的腦組織和AD小鼠模型�����,通過免疫組織化學(xué) (IHC) 染色和免疫印跡法測定壞死信號(hào)通路中關(guān)鍵基因的表達(dá)�����,發(fā)現(xiàn)TNF-α/TNFR1信號(hào)通路在AD神經(jīng)元壞死激活中發(fā)揮作用�。在TNF-α刺激下��,積累的p62 招募 RIPK1并誘導(dǎo)其自我寡聚,激活下游 RIPK1/RIPK3/MLKL級(jí)聯(lián)反應(yīng)�����,導(dǎo)致神經(jīng)元壞死���。進(jìn)一步研究發(fā)現(xiàn),p62的異常積累是由TNF-α誘導(dǎo)的UVRAG下調(diào)介導(dǎo)的自噬損傷引起的���。通過注射AAV載體上調(diào)UVRAG的表達(dá),可以抑制 AD細(xì)胞和小鼠模型中神經(jīng)元壞死����,并改善認(rèn)知功能�。本研究揭示了AD發(fā)病過程中神經(jīng)元壞死和自噬機(jī)制之間的聯(lián)系��,為AD的臨床干預(yù)提供了潛在的靶點(diǎn)���,同時(shí)也為其他NDs的細(xì)胞死亡調(diào)節(jié)機(jī)制的探索提供了基礎(chǔ)。
為探究AD患者腦內(nèi)TNF-α水平的升高是否會(huì)導(dǎo)致神經(jīng)元的壞死����,作者檢測了AD患者和小鼠的腦組織中MLKL和p-MLKL(壞死標(biāo)志物)的表達(dá)�����,發(fā)現(xiàn)在AD中細(xì)胞壞死確實(shí)被激活����。通過在小鼠側(cè)腦室注射TNF-α����,發(fā)現(xiàn)高水平的TNF-α可以增加p-MLKL的表達(dá)并激活壞死����。使用AAV9對(duì)TNFR1(TNF-α受體)進(jìn)行敲低,發(fā)現(xiàn)TNFR1敲低后能導(dǎo)致p-MLKL表達(dá)水平的降低�����,提示TNF-α/TNFR1信號(hào)通路在AD神經(jīng)元壞死過程中發(fā)揮重要作用。
圖2. TNF-α/TNFR1誘導(dǎo)神經(jīng)元壞死
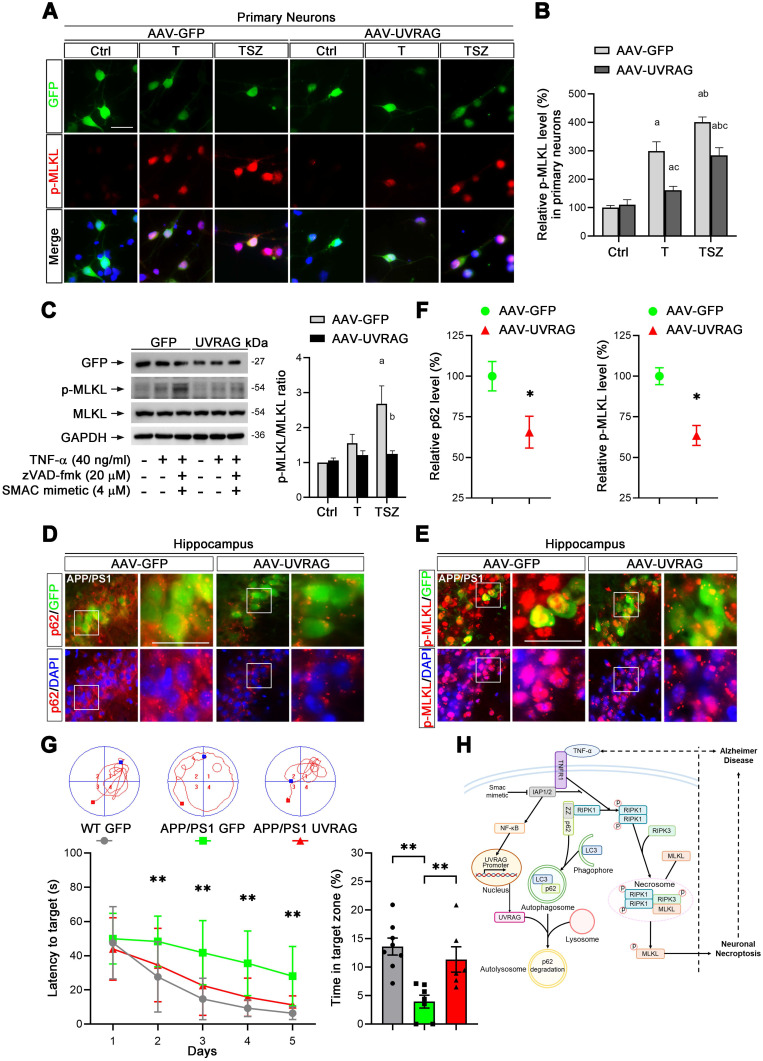
研究人員發(fā)現(xiàn)在TNF-α誘導(dǎo)下�����,p62積累并激活下游 RIPK1/RIPK3/MLKL級(jí)聯(lián)反應(yīng)�����,導(dǎo)致了神經(jīng)元壞死,而p62的異常積累是由UVRAG下調(diào)介導(dǎo)的自噬流受損引起的�。為進(jìn)一步探究UVRAG在AD小鼠神經(jīng)元壞死中的作用���,研究者利用AAV載體在原代神經(jīng)元中對(duì)UVRAG進(jìn)行了過表達(dá)�����,結(jié)果發(fā)現(xiàn)UVRAG的過表達(dá)明顯降低了TNF-α誘導(dǎo)的MLKL磷酸化。通過在AD小鼠海馬區(qū)注射AAV-UVRAG�,發(fā)現(xiàn)p62與MLKL的磷酸化水平均顯著降低���。此外,UVRAG的過表達(dá)明顯改善了AD小鼠的學(xué)習(xí)和記憶功能障礙���。以上結(jié)果表明���,上調(diào)UVRAG可抑制AD小鼠神經(jīng)元壞死并改善認(rèn)知功能,提示UVRAG可能是AD干預(yù)的潛在靶點(diǎn)��。
圖3. UVRAG抑制原代神經(jīng)元和AD小鼠中的神經(jīng)元壞死
· 案例二 ·
“Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease”
帕金森病(PD)是常見的神經(jīng)退行性疾病�����,已有研究表明���,α-突觸蛋白(α-syn)誘導(dǎo)的小膠質(zhì)細(xì)胞活化是PD發(fā)病的重要因素之一��,代謝型谷氨酸受體5(mGluR5)及其信號(hào)通路在保護(hù)神經(jīng)元免受神經(jīng)炎癥中發(fā)揮重要作用,且mGluR5可能在α-SYN誘導(dǎo)的PD神經(jīng)炎癥和神經(jīng)毒性的發(fā)病機(jī)制中起重要作用��,但其具體調(diào)控機(jī)制尚不明確��。為探討mGluR5在α-SYN誘導(dǎo)的小膠質(zhì)細(xì)胞炎癥中的作用和機(jī)制��,作者通過過表達(dá)α-SYN來激活小膠質(zhì)細(xì)胞��,發(fā)現(xiàn)mGluR5的激活部分抑制了α-SYN誘導(dǎo)的炎癥反應(yīng)����,保護(hù)小膠質(zhì)細(xì)胞免受神經(jīng)毒性。此外��,α-SYN通過與溶酶體結(jié)合來促進(jìn)mGluR5的降解機(jī)制在PD動(dòng)物模型中同樣存在���。這些發(fā)現(xiàn)為解密mGluR5調(diào)控α-SYN誘導(dǎo)的小膠質(zhì)細(xì)胞炎癥機(jī)制���,探究PD未知的發(fā)病機(jī)制提供了新的思路����。

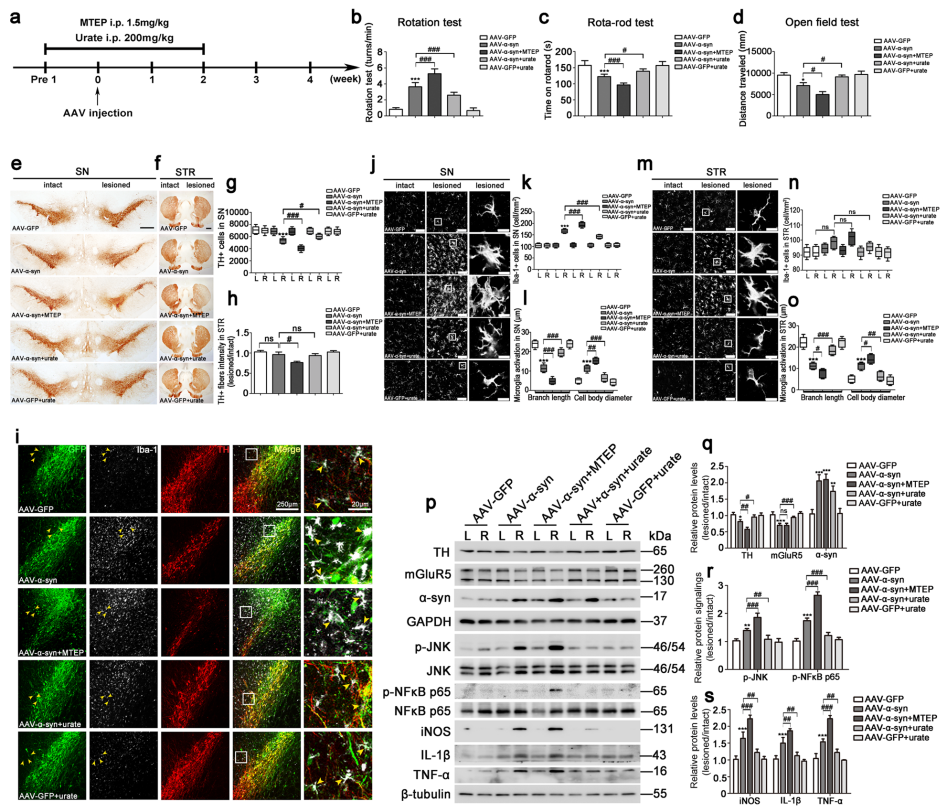
為確定mGluR5在α-syn誘導(dǎo)的體內(nèi)小膠質(zhì)細(xì)胞激活中的功能作用,作者利用AAV9-α-syn誘導(dǎo)PD大鼠模型�����,并對(duì)其進(jìn)行行為評(píng)估及病理生理學(xué)分析,發(fā)現(xiàn)α-syn過表達(dá)組從第8周至第16周出現(xiàn)顯著的進(jìn)行性功能障礙�����,且過表達(dá)α-syn��,能導(dǎo)致mGluR5表達(dá)降低��,同時(shí)TH表達(dá)改變;此外小膠質(zhì)細(xì)胞中的炎性因子iNOS����、IL-1β和TNF-α的表達(dá)水平在注射后10天開始升高,4周時(shí)達(dá)到峰值�����,8周后影響消失�����,這說明在PD的發(fā)病早期伴隨著反應(yīng)性小膠質(zhì)細(xì)胞的高度局部炎癥反應(yīng)��。為進(jìn)一步確定炎癥干預(yù)在PD早期的作用��,作者在第4周炎癥高峰期時(shí)利用MEPT(mGluR5拮抗劑)或尿酸鹽處理��,結(jié)果發(fā)現(xiàn)MEPT處理阻斷了mGluR5的活性并加劇了炎癥反應(yīng),而尿鹽酸處理減緩了炎癥反應(yīng)���。綜上����,在AAV-α-syn誘導(dǎo)的PD模型中����,mGluR5的激活在介導(dǎo)神經(jīng)保護(hù)的抗炎反應(yīng)中起著關(guān)鍵作用。
圖4. mGluR5在AAV-α- sync誘導(dǎo)的大鼠PD模型中具有抗炎作用
· 案例三 ·
“PHLDA1 promotes microglia-mediated neuroinflammation via regulating K63-linked ubiquitination of TRAF6”
PD作為一種常見的年齡相關(guān)性神經(jīng)退行性疾病��,以黑質(zhì)致密部(SNc)中多巴胺能(DA)神經(jīng)元的進(jìn)行性丟失為特征�����。小膠質(zhì)細(xì)胞介導(dǎo)的神經(jīng)炎癥在PD等神經(jīng)退行性疾病的進(jìn)展中發(fā)揮著重要作用�����。目前普遍認(rèn)為��,TLR4/TRAF6/NF-κB或MAPKs信號(hào)通路在小膠質(zhì)細(xì)胞活化介導(dǎo)的神經(jīng)炎癥的調(diào)節(jié)中發(fā)揮關(guān)鍵作用��。已知PHLDA1(人Plecstrin同源域家族A成員 1,也稱T細(xì)胞死亡相關(guān)基因51�,TDAG51)參與了對(duì)TLR(Toll樣受體)激活的免疫反應(yīng)的調(diào)節(jié),然而���,PHLDA1在免疫應(yīng)答中的確切分子信號(hào)和功能作用尚未完全闡明�,特別是在小膠質(zhì)細(xì)胞激活和神經(jīng)退行性疾病中的功能作用尚不清楚����。
研究人員發(fā)現(xiàn)在體內(nèi)和體外小膠質(zhì)細(xì)胞中�����,PHLDA1的表達(dá)在炎癥刺激下迅速增加����。利用腺相關(guān)病毒(AAV)下調(diào)PHLDA1可改善MPTP誘導(dǎo)的小鼠運(yùn)動(dòng)缺陷并抑制神經(jīng)炎癥�。在體內(nèi)�����,發(fā)現(xiàn)LPS誘導(dǎo)的TNF-α��、IL-1β�、iNOS和COX-2等促炎基因表達(dá)在PHLDA1敲除的小膠質(zhì)細(xì)胞中降低����。機(jī)制研究表明,LPS刺激后�,小膠質(zhì)細(xì)胞中PHLDA1表達(dá)增加,導(dǎo)致與TRAF6的直接相互作用����,并增強(qiáng)其K63連接的泛素化介導(dǎo)的NF-κB信號(hào)通路激活。PHLDA1缺乏可干擾TRAF6 k63的泛素化并抑制小膠質(zhì)細(xì)胞炎癥反應(yīng)�。本研究表明了PHLDA1與小膠質(zhì)細(xì)胞介導(dǎo)的DA神經(jīng)毒性有關(guān)�,提示PHLDA1可能是一種有效的神經(jīng)炎癥調(diào)節(jié)劑���,為治療神經(jīng)炎癥相關(guān)疾病(如PD)提供新的藥物靶點(diǎn)。

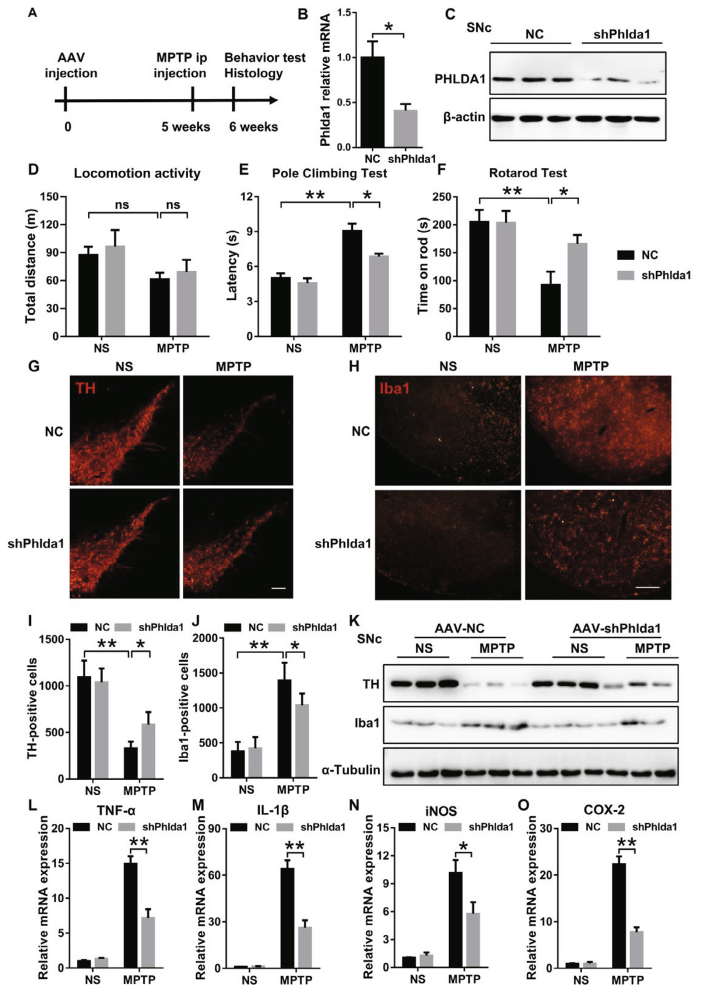
為探究PHLDA1在PD中的作用����,研究人員利用AAV介導(dǎo)的shRNA對(duì)小鼠體內(nèi)PHLDA1進(jìn)行了敲降�����,5周后檢測發(fā)現(xiàn)AAV9-shPhlda1組小鼠���,PHLDA蛋白與mRNA水平均顯著下降���。感染小鼠給予MPTP誘導(dǎo)PD樣表型,發(fā)現(xiàn)抑制PHLDA1的表達(dá)無法改善小鼠基礎(chǔ)運(yùn)動(dòng)功能�����,但能顯著減弱MPTP誘導(dǎo)的行為損傷��,對(duì)ShPhlda1小鼠中腦的TH免疫組化染色和免疫印跡進(jìn)一步證實(shí)了其保護(hù)作用���,這表明抑制PHLDA1的表達(dá)可顯著減輕MPTP所致的多巴胺能神經(jīng)元和行為損傷���。有趣的是�,ShPhlda1小鼠SNc中Iba-1(小膠質(zhì)細(xì)胞標(biāo)志物)陽性細(xì)胞數(shù)量和總蛋白水平也降低,且ShPhlda1小鼠中炎性因子的表達(dá)降低�。這些結(jié)果表明,SNc中PHLDA1的耗竭保護(hù)了DA神經(jīng)元并抑制了神經(jīng)炎癥�����,最終減弱了MPTP誘導(dǎo)的PD樣表型的發(fā)展�。
圖5. SNc中PHLDA1敲除可顯著改善MPTP誘導(dǎo)的DA神經(jīng)元損傷和運(yùn)動(dòng)障礙
· 案例四 ·
“Upregulation of KDM6B contributes to lipopolysaccharide-induced anxiety-like behavior via modulation of VGLL4 in mice ”
神經(jīng)精神障礙如焦慮癥,近年來已發(fā)展成為日益嚴(yán)重的公共心理健康問題����。有證據(jù)表明,LPS誘導(dǎo)的神經(jīng)炎癥與焦慮樣行為之間存在因果關(guān)系�����,阻斷神經(jīng)炎性介質(zhì)已被認(rèn)為是治療焦慮的一種潛在方法����。近年來�����,表觀遺傳學(xué)被認(rèn)為在神經(jīng)精神障礙如焦慮癥中起著至關(guān)重要的作用����,其中組蛋白H3K27me3去甲基化酶KDM6B作為一個(gè)關(guān)鍵的表觀遺傳調(diào)節(jié)因子��,在炎癥���、細(xì)胞分化�����、神經(jīng)再生、癌癥以及中樞神經(jīng)系統(tǒng)和行為反應(yīng)等中發(fā)揮重要作用�����。然而��,KDM6B在神經(jīng)炎癥誘發(fā)的焦慮樣行為中的作用知之甚少��。本研究探討了KDM6B在LPS誘導(dǎo)的焦慮樣行為中的潛在作用�,并評(píng)估了其是否與VGLL4的調(diào)控有關(guān)。

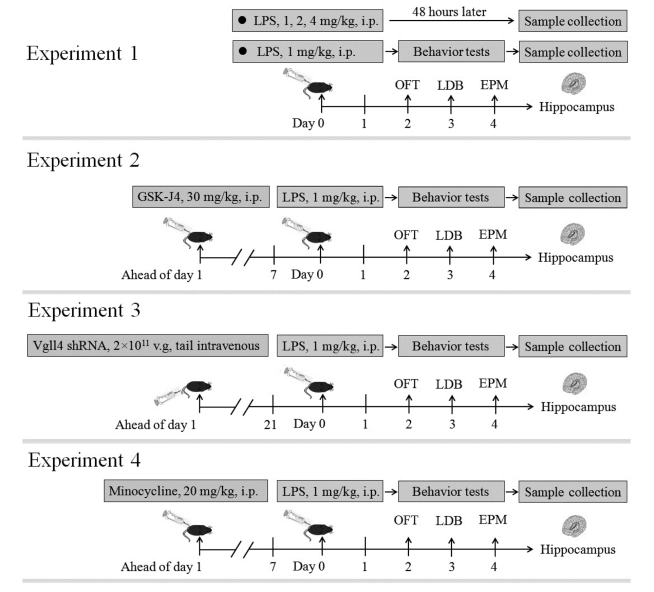
首先���,研究者使用LPS誘導(dǎo)小鼠焦慮樣行為,并測定小鼠海馬區(qū)和前額皮質(zhì)中KDM6B和VGLL4的表達(dá)以及小鼠海馬區(qū)焦慮樣行為和神經(jīng)炎癥的變化�,發(fā)現(xiàn)海馬區(qū)KDM6B、VGLL4�、IL-1β和Iba-1的表達(dá)水平呈LPS劑量依賴式增加��,并伴隨焦慮樣行為的增加�。接著����,分析了KDM6B抑制劑GSK-J4對(duì)LPS誘導(dǎo)的焦慮樣行為的改變,發(fā)現(xiàn)GSK-J4 處理減弱了LPS誘導(dǎo)的 VGLL4���、STAT3�、IL-1β 和 Iba-1的上調(diào)���,并減輕了焦慮樣行為����。隨后����,用AAV介導(dǎo)的VgLL4 shRNA 敲低VGLL4�,阻止了LPS誘導(dǎo)的小鼠海馬區(qū)焦慮樣行為和STAT3���、IL-1β 和 Iba-1 表達(dá)水平的增加�����。最后����,發(fā)現(xiàn)小膠質(zhì)細(xì)胞抑制劑-米諾環(huán)素(MNC)可以減弱LPS誘導(dǎo)的焦慮樣行為��。本研究發(fā)現(xiàn)�,LPS誘導(dǎo)的神經(jīng)炎癥促進(jìn)了海馬區(qū)中KDM6B 的激活,并且LPS誘導(dǎo)的焦慮樣行為與海馬區(qū)中KDM6B誘導(dǎo)VGLL4的上調(diào)有關(guān)�����。
圖6. 研究思路
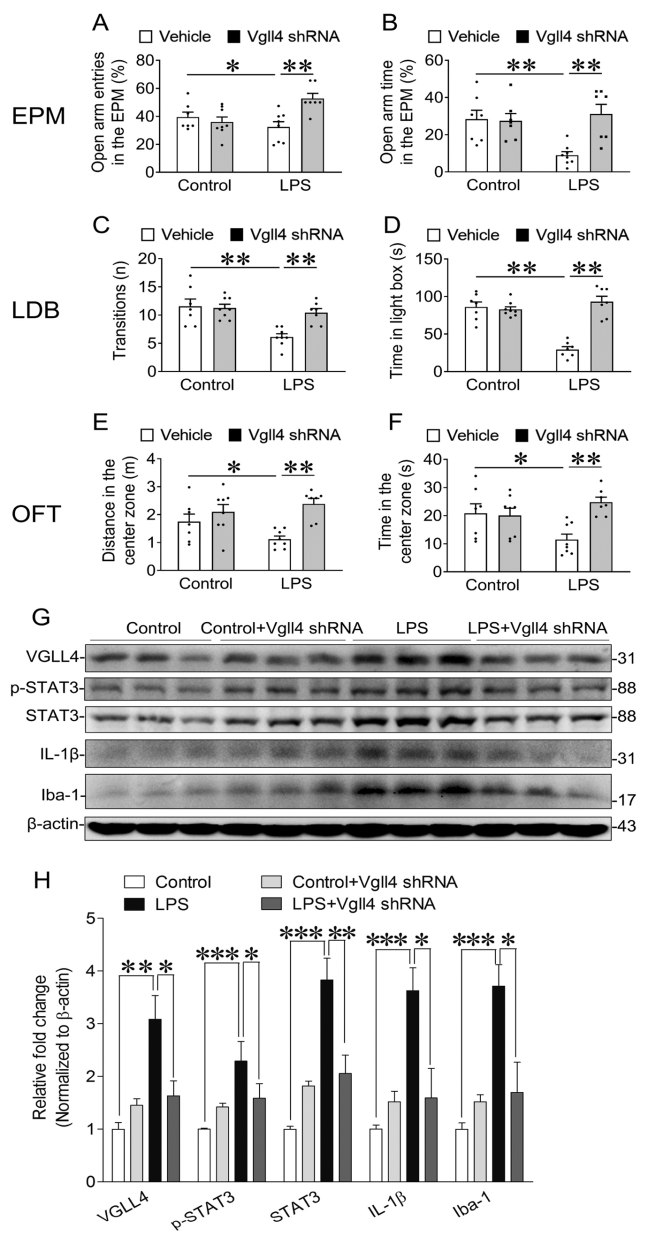
為進(jìn)一步探究VGLL4在LPS誘導(dǎo)的焦慮樣行為中的作用�����,作者在LPS注射前����,使用AAV介導(dǎo)的VGLL4 shRNA對(duì)VGLL4進(jìn)行敲低處理�����,通過曠場�����、迷宮等行為學(xué)測試發(fā)現(xiàn)��,敲低VGLL4可以減輕LPS誘導(dǎo)的小鼠焦慮樣行為�����。此外���,LPS誘導(dǎo)了海馬區(qū)STAT3(信號(hào)轉(zhuǎn)導(dǎo)和轉(zhuǎn)錄激活因子3)表達(dá)的增加,而這種增加在敲低VGLL4后被逆轉(zhuǎn)�����,并且VGLL4的敲低也顯著減弱了LPS誘導(dǎo)的海馬區(qū)IL-1β和Iba-1的表達(dá)�����。這些研究結(jié)果表明,VGLL4和海馬中的STAT3之間存在促進(jìn)關(guān)系���,VGLL4通過上調(diào)海馬中的STAT3來促進(jìn)LPS誘導(dǎo)的焦慮樣行為。
圖7. VGLL4敲低對(duì)LPS誘導(dǎo)的焦慮樣行為和海馬中相關(guān)蛋白表達(dá)水平的影響