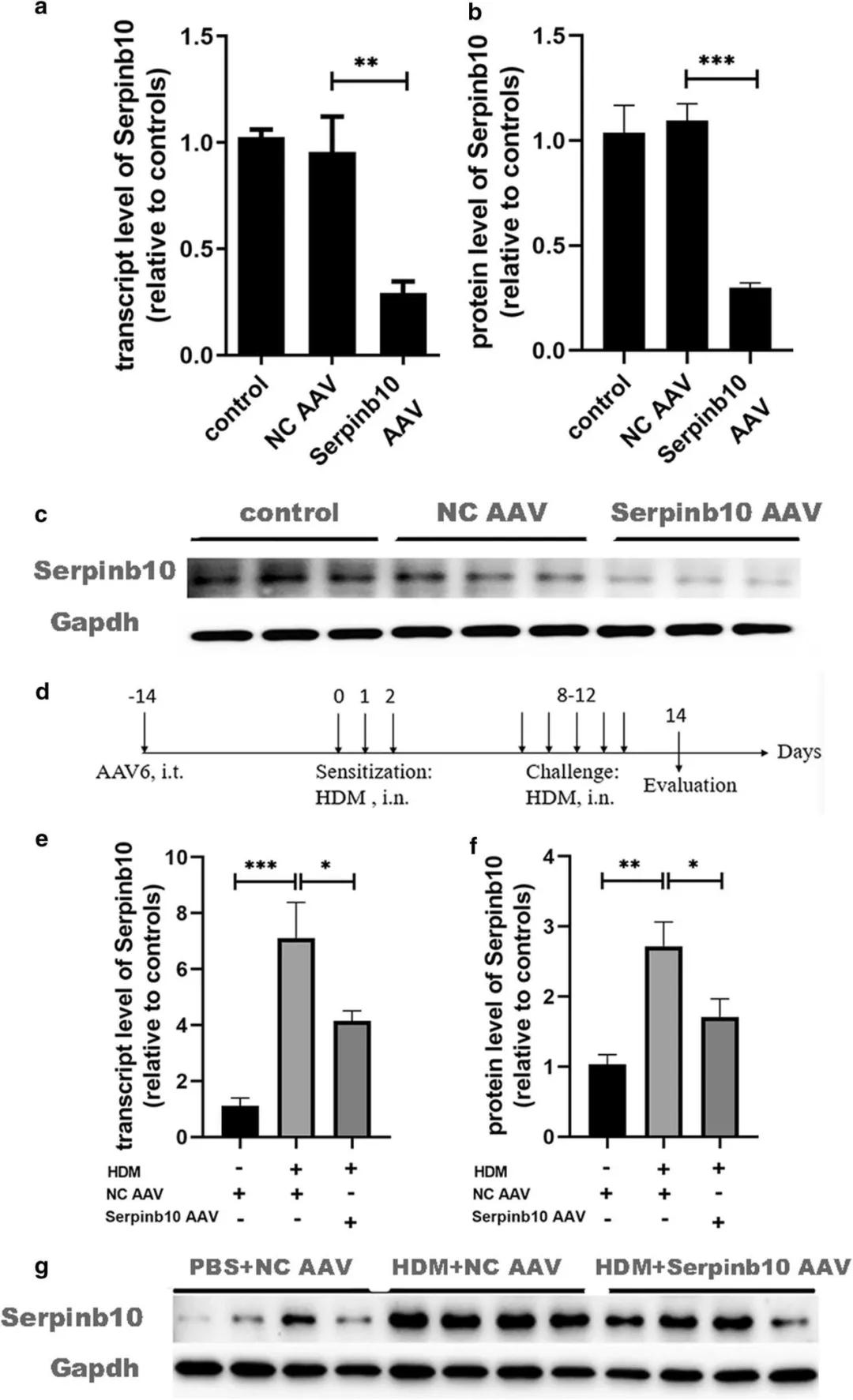
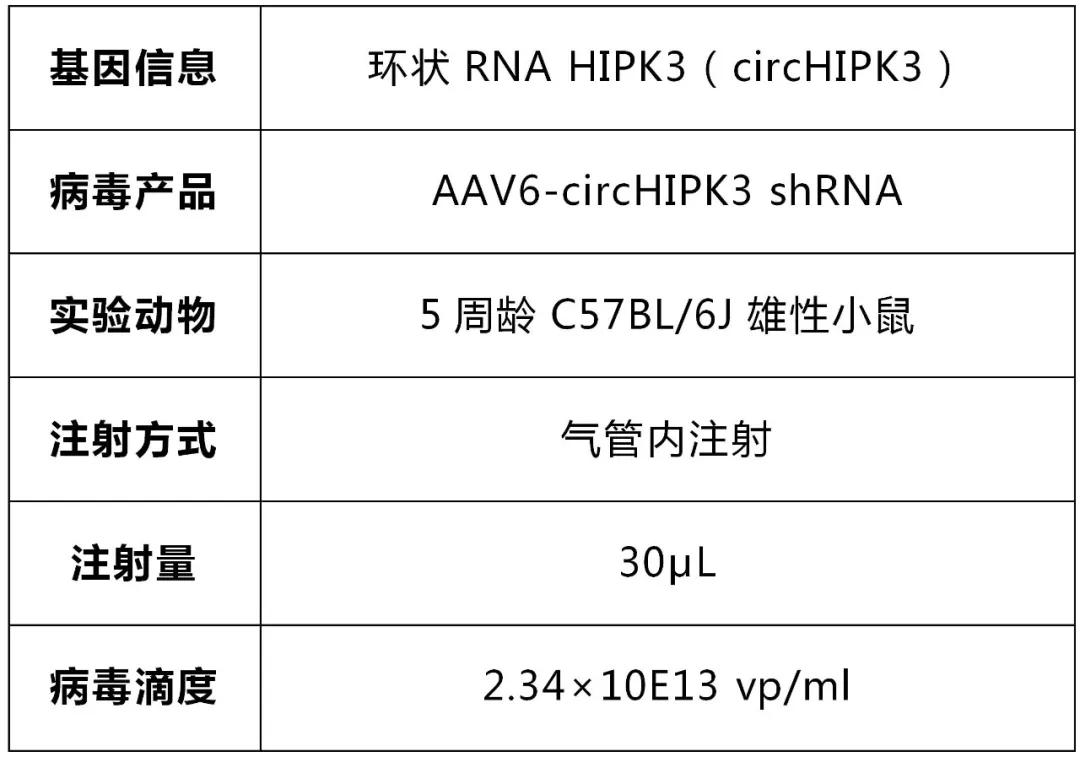
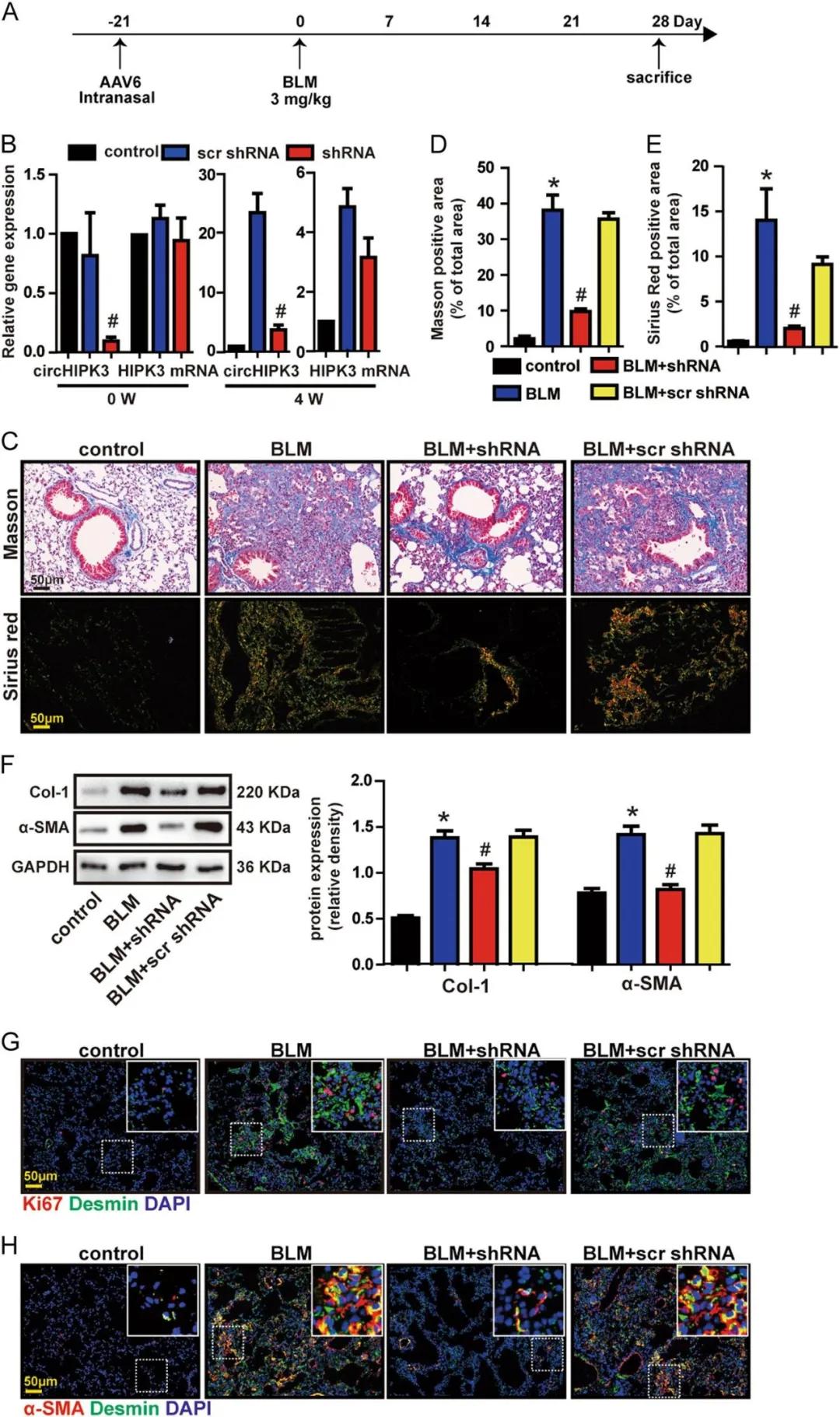
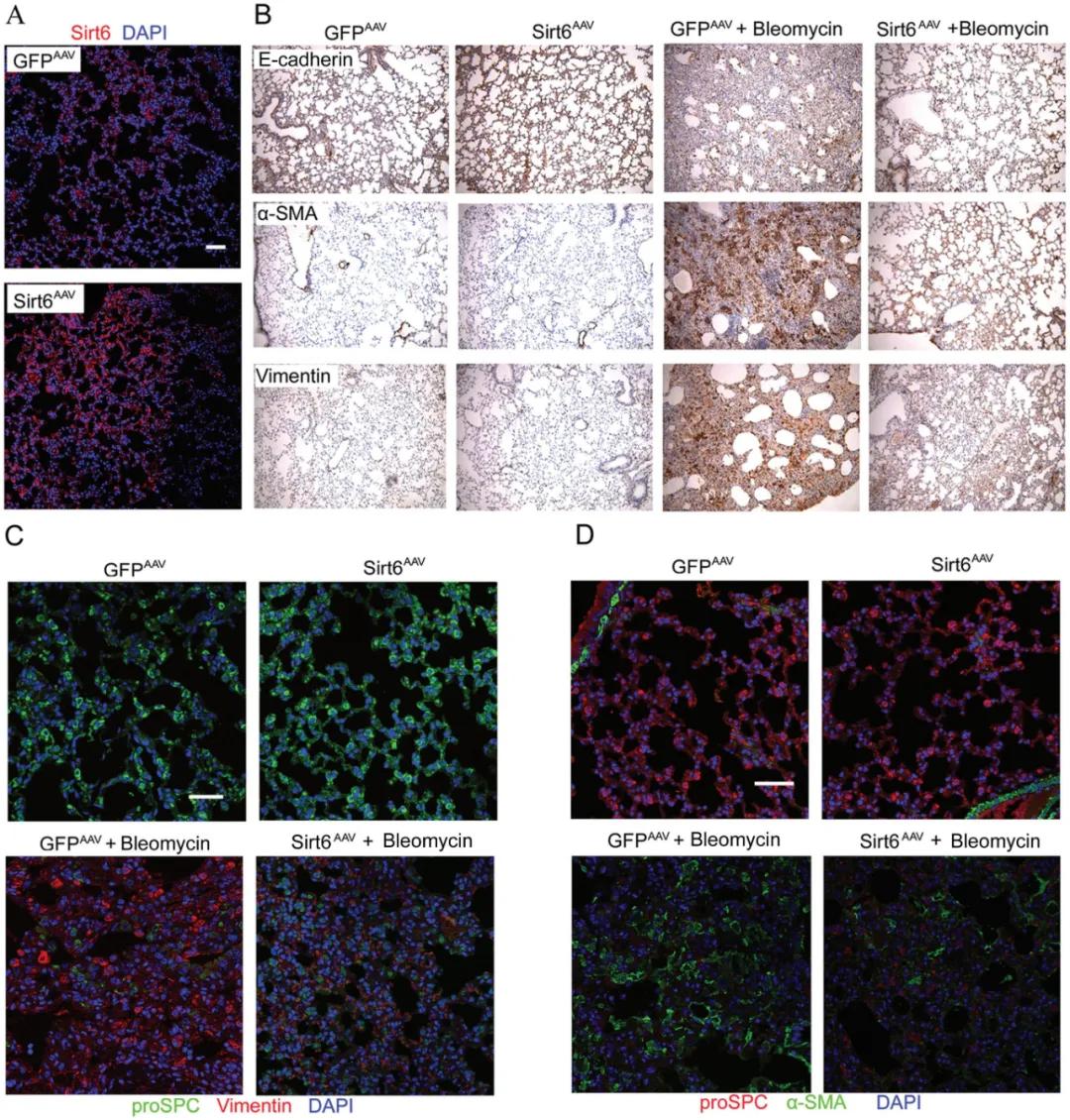
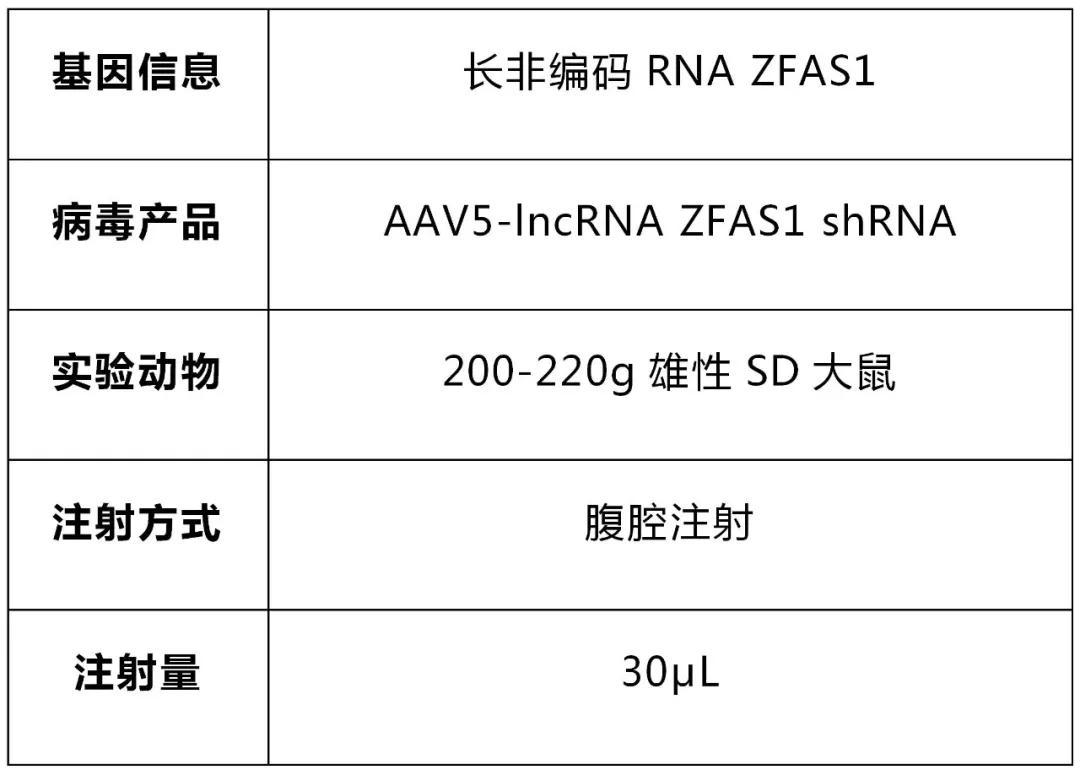
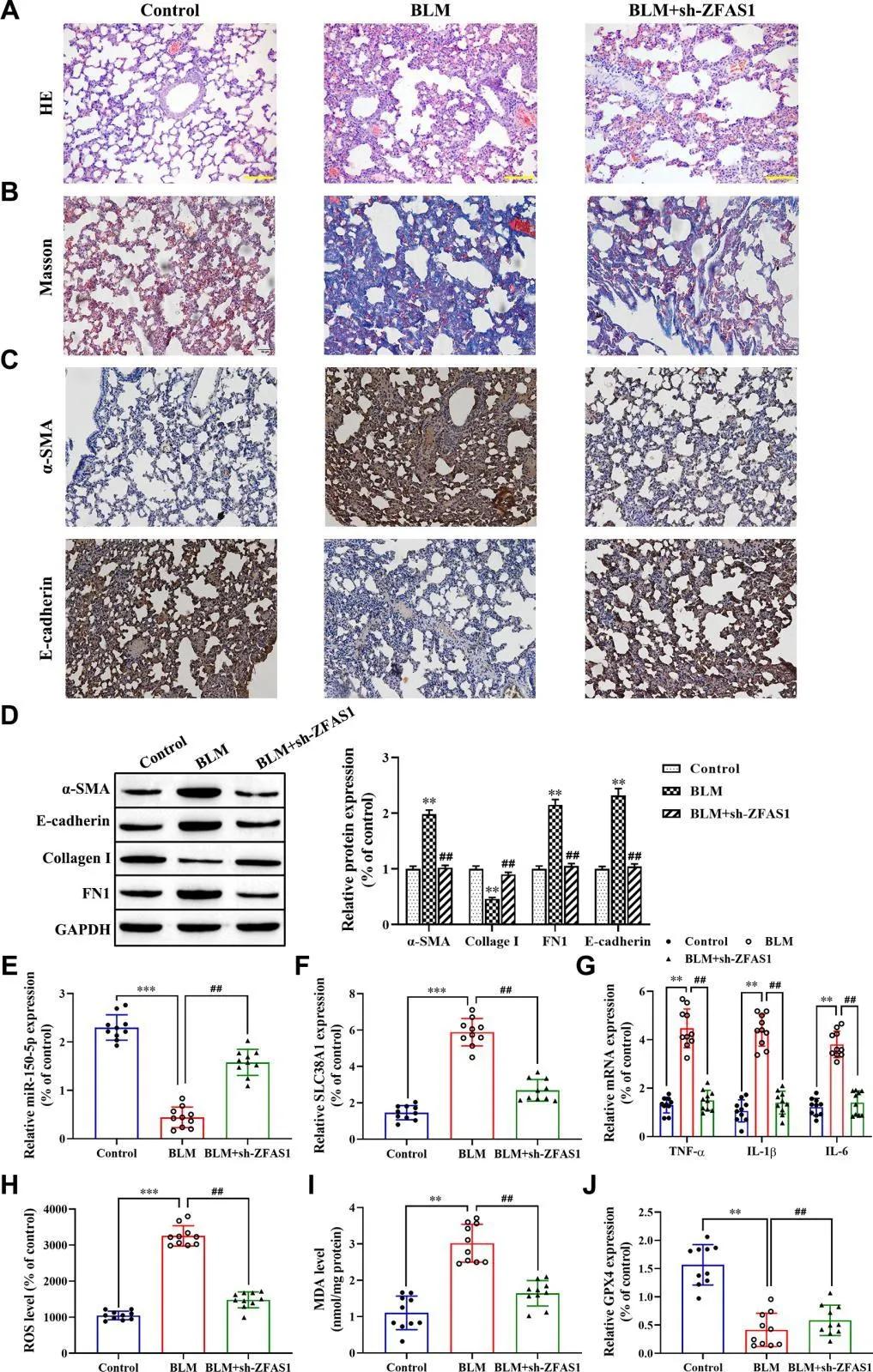
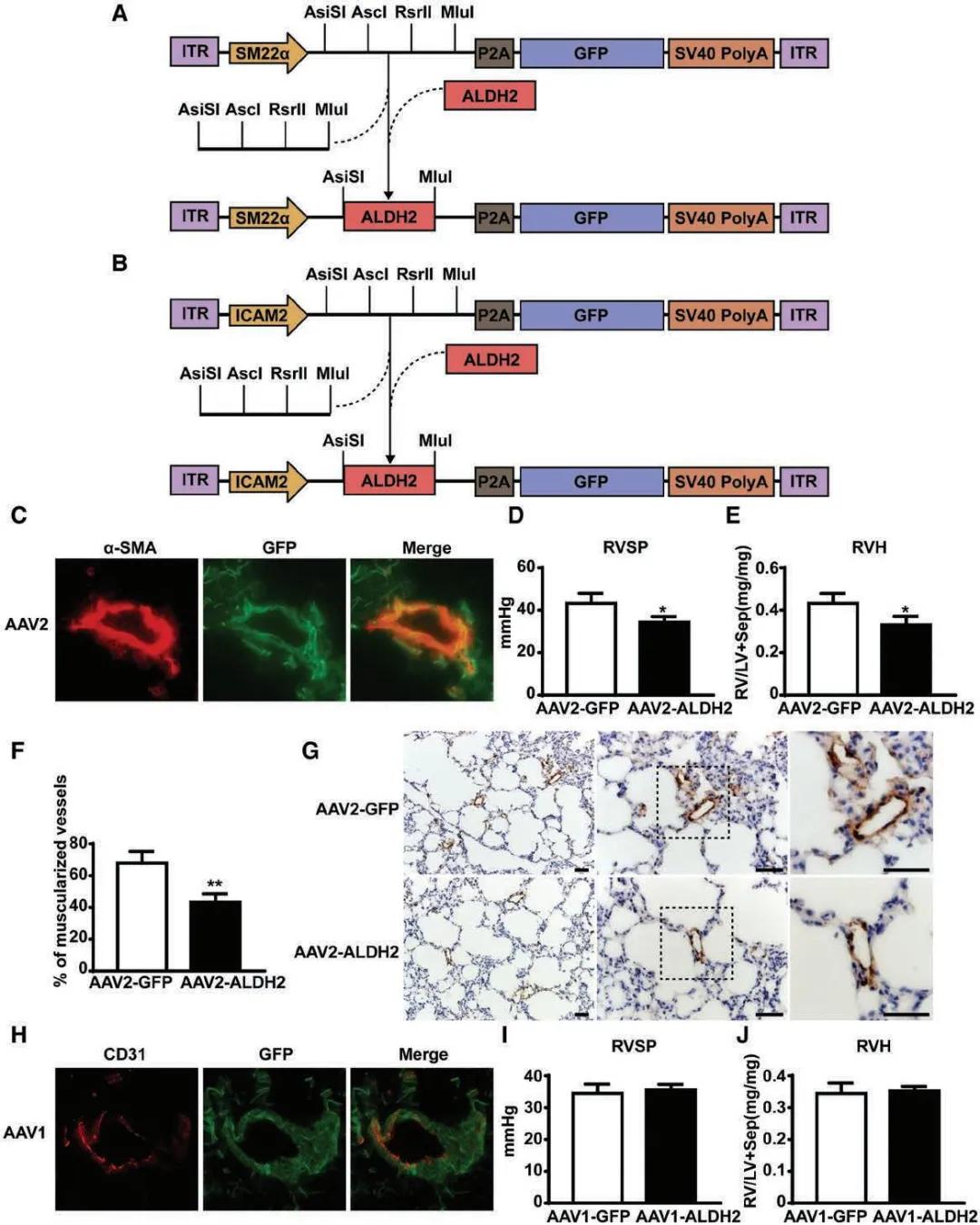
請選擇
- 全部
- 質(zhì)粒-人源cDNA
- 質(zhì)粒-人源miRNA
- 質(zhì)粒-人源shRNA
- 質(zhì)粒-gRNA
- 質(zhì)粒-人源cDNA慢病毒
- 質(zhì)粒-小鼠cDNA
- 維真質(zhì)粒庫
- 腺病毒-人源cDNA
- 腺病毒-人源miRNA
- 常備現(xiàn)貨腺病毒
- 現(xiàn)貨腺相關(guān)病毒
- 現(xiàn)貨慢病毒
- 預(yù)制敲除細(xì)胞系